Chief
 |
OTANI Minoru, Professor
He graduated from Osaka University in 2000, with a doctor degree. Following his graduation, he initially served as a postdoctoral fellow of Japan Society for the Promotion Science working at the University of Tokyo. He served as a research associate at the University of Tsukuba (2001), a research associate at the University of Tokyo (2003), a researcher (2008), a senior researcher (2009) and a group leader (2010) in the National Institute of Advanced Industrial Science and Technology (AIST). Since 2021, he has joined the Center for Computational Sciences (CCS) in the University of Tsukuba as a full professor. His major is computational material science. |
Overview
All the materials around us are composed of atoms, and atoms are composed of nuclei and electrons. Materials exhibit various properties reflecting their composition and structures and they are widely used in today’s science and technology. In this division, we study quantum many-body systems–the substances coupled by Coulomb interaction–by solving quantum mechanical equations of motion numerically. Our goals are to elucidate various properties of materials, to find devices of new functions, and to search a way to control the dynamical processes. Such a research is the foundation for the future technology.
Research topics
Light-matter interactions
Lights have been used to measure the properties of materials accurately. Recently, in optical sciences, a strong ultrashort pulsed light is used to measure the fast electronic motion in real time and to control the electronic motion. We develop a simulation code based on the first-principles calculations such as the time-dependent density functional theory to investigate the electronic dynamics and to understand the mechanism of the light-matter interactions.
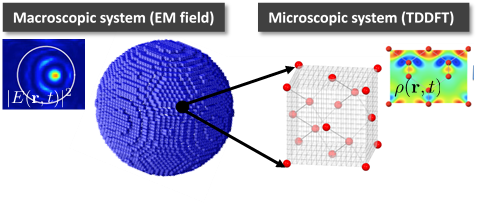 Development of first-principles, real-time and real-space simulation code for optical science
Development of first-principles, real-time and real-space simulation code for optical science
In current frontiers of optical science, innovative simulation methods describing light-matter interaction from microscopic electronic dynamics are required. As such computer code, we develop a first-principles computational code SALMON (Scalable Ab-initio Light-Matter simulator for Optics and Nanoscience) that is based on density functional theory and utilizes real-space and real-time method, under close collaborations with computer scientists in the Center and researchers in physics and optics world-wide. SALMON is developed as an open-source software, and can be downloaded from the website, https://salmon-tddft.jp.
 Strongly correlated materials and topological materials
Strongly correlated materials and topological materials

Strongly correlated materials or topological materials whose properties depend on the shape of the material exhibit interesting properties that cannot be explained by the band theory. We develop a numerical method to study properties of these systems including electronic states, phase transitions, optical responses, and perform a research toward developing new quantum information devices.
Web sites
KOIZUMI Laboratory (Japanese)
(Update: Dec. 18, 2019)